
{kind=link}
Porcine reproductive and respiratory syndrome virus (PRRSV) together with African swine fever virus is without doubt one of the most economically necessary pathogens affecting the swine trade globally. The latest emergence of aggressive PRRSV strains just like the Extremely Pathogenic PRRSV (HP-PRRSV) in Asia, Rosalia in Europe, and the L1C.5 in North America has ignited the dialogue on the necessity to additional enhance PRRSV diagnostics and management.
Using RT-PCR for detecting PRRSV genetic materials is often used worldwide to display populations for the virus.
One step past RNA detection is PRRSV genetic sequencing, generally finished utilizing the Sanger method. Globally, the portion of the virus mostly used for PRRSV sequencing is ORF5, though some laboratories report ORF7 sequencing.
The ORF5 represents about 4% and ORF7 2.4% of the PRRSV genome, and thus, these don’t present full genetic protection of the entire genome.
In recent times, there was a rising curiosity in using next-generation sequencing (NGS) for the restoration of complete PRRSV genomes for PRRSV epidemiological investigations inside a herd or manufacturing system (Determine 1).
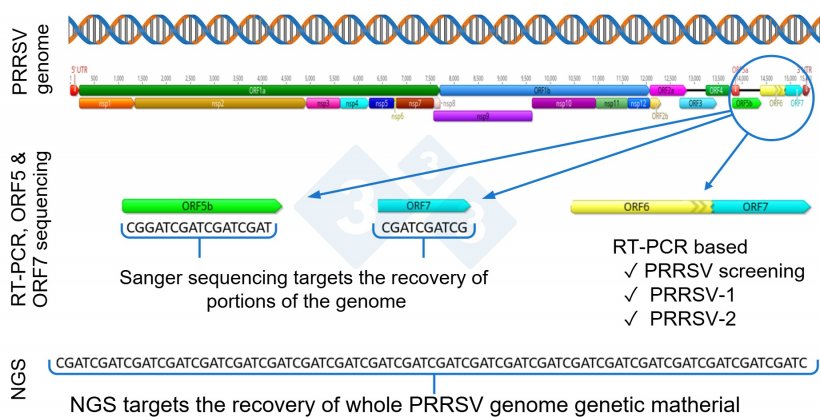
Usually, in the course of the replication course of, PRRSV goes by means of genetic adjustments and mutations, that will doubtlessly result in the emergence of recent viral variants. PRRSV is without doubt one of the viruses identified to have a excessive price of mutations, about 0.5 to 1% a 12 months, erratically distributed throughout totally different genome areas, virus kind, and genetic lineages, resulting in fixed genetic evolution. Notably, genetic mutation and evolution of the virus can happen throughout all genes, and sequencing solely a portion of the genome, e.g., ORF5 or ORF7, seemingly misses the chance to detect adjustments that occurred exterior the sequenced area (Determine 1).
Beneath this state of affairs, NGS turns into a great tool by offering a possibility to get well an entire PRRSV genome for use in epidemiological investigations.
How can veterinarians and producers make the benefit of NGS
To maximise the utility of NGS, there are some factors to think about:
- Early involvement of the veterinarian and laboratory diagnostician is necessary to align the diagnostic query, expectations, and strategy for testing.
- Is there a PRRSV referent pressure complete genome sequence on a farm or manufacturing system for comparability?
- If not, use NGS on PRRSV RT-PCR optimistic samples with low Ct values, i.e., ideally < 24, to get well an entire genome because the herd-referent pressure. The Ct worth is inversely associated to the quantity of genetic copies of the virus current within the pattern, i.e., the decrease the Ct, the upper the anticipated viral load and potential success of NGS;
- A referent pressure permits subsequent comparability of prospectively recovered genomes to know PRRSV genetic evolution throughout the farm, stream, and system.
- What’s the goal of doing NGS?
- If the target is to detect virus evolution on the complete genome stage: goal lung and serum since these samples usually tend to get well an entire genome.
- If the target is to know the viral range in a farm or herd, use inhabitants pattern varieties like processing fluid, oral fluid samples, or pooled particular person samples, e.g., pooled serum is extra vulnerable to get well a number of viruses if current within the pattern.
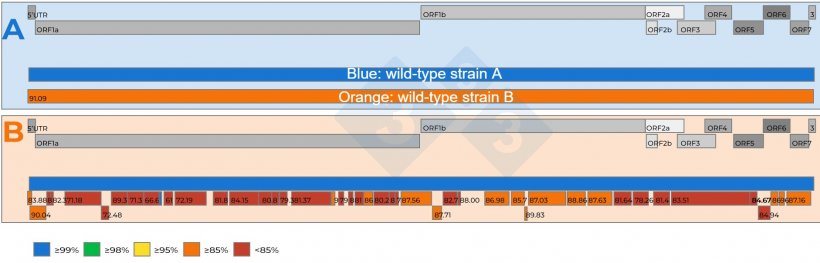
- Combined infections with two or extra PRRSVs in a herd are a actuality (Determine 2a), and if the 2 viruses are very related, the NGS could fail to get well an entire genome;
- If a number of viruses are current within the pattern, NGS could get well genome fragments, known as ‘contigs’, from totally different viruses, which, when in comparison with a farm reference pressure, will help discern if a number of viruses are current within the pattern (determine 2b);
- Lastly, be sure you have somebody to assist with the genetic evaluation and comparisons, and coordinate the turnaround time, as NGS is often a gradual course of that may take over every week to finish.
Which sort of epidemiological info can we get from NGS
Generated NGS outputs will help type out questions:
- Did a virus evolve by means of random nucleotide substitutions on the similar genomic positions? How a lot has the virus modified between two time factors, and wherein genome, i.e., ORF area(s), did these adjustments happen?
- Did the virus evolve by means of insertions or deletions in its genome?
- Did a brand new introduction of an unrelated virus happen within the farm, herd, or stream?
- Though much less seemingly however potential, did a brand new virus acquired adjustments within the genomic areas focused by RT-PCR or Sanger primer/probe sequencing, inflicting the assays to fail in detection?
- Did the virus endure recombination, i.e., acquired some genomic areas from two or extra parental viruses
Recombination is a pure means of PRRSV evolution and happens when two PRRSV replicate in the identical cell producing a 3rd derived virus. Recombination will likely be additional explored in article revealed on pig333.com on 9 June 2025 “The implications of the PRRSV recombination dilemma”.